
Definition
- The urea cycle:
- Is the sole source of endogenous production of arginine, ornithine, and citrulline;
- Is the principal mechanism for the clearance of waste nitrogen resulting from protein turnover;
- Is the principal mechanism for the metabolism of other nitrogenous metabolic compounds such as adenosine monophosphate;
- Includes enzymes that overlap with the nitric oxide production pathway (ASS1 and ASL).
The urea cycle comprises the following components:
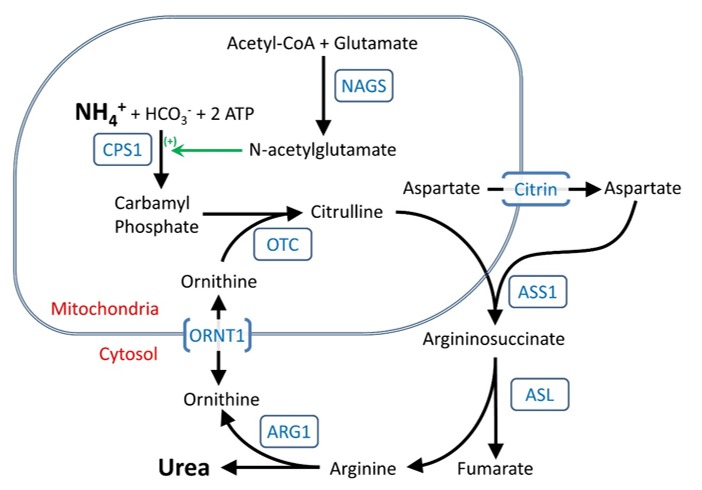
- Five catalytic enzymes:
- Carbamoylphosphate synthetase I (CPS1)
- Ornithine transcarbamylase (OTC)
- Argininosuccinic acid synthetase (ASS1)
- Argininosuccinic acid lyase (ASL)
- Arginase (ARG1)
- One cofactor-producing enzyme:
- N-acetyl glutamate synthetase (NAGS)
- Two amino acid transporters:
- Ornithine translocase (ORNT1; ornithine/citrulline carrier; solute carrier family 25, member 15)
- Citrin (aspartate/glutamate carrier; solute carrier family 25, member 13)
Clinical Characteristics of Urea Cycle Disorders (UCDs)
- Urea cycle disorders (UCDs) result from inherited deficiencies in any one of the six enzymes or two transporters of the urea cycle pathway, specifically:
- CPS1 (Carbamoylphosphate synthetase I)
- OTC (Ornithine transcarbamylase)
- ASS1 (Argininosuccinic acid synthetase)
- ASL (Argininosuccinic acid lyase)
- ARG1 (Arginase)
- NAGS (N-acetyl glutamate synthetase)
- ORNT1 (Ornithine translocase)
- Citrin (Aspartate/glutamate carrier)
Clinical Characteristics of Urea Cycle Disorders
- The severity of urea cycle defects is influenced by:
- The position of the defective protein in the pathway
- The severity of the enzymatic deficiency
Severe Urea Cycle Defects (Neonatal Onset)
- Severe deficiency or total absence of activity of CPS1, OTC, ASS1, ASL, or NAGS causes rapid ammonia accumulation due to the lack of effective alternative clearance mechanisms.
- Presentation:
- Typically occurs in the neonatal period due to liver immaturity
- Infants appear normal at birth but rapidly develop cerebral edema leading to symptoms such as:
- Lethargy, anorexia
- Hyperventilation or hypoventilation
- Hypothermia
- Seizures, neurological posturing
- Coma
- Initial nonspecific symptoms:
- Failure to feed
- Loss of thermoregulation (hypothermia)
- Somnolence progressing to lethargy and coma
- Approximately 50% may experience seizures.
- Closed cranial sutures increase risk of rapid neurological deterioration.
- Respiratory changes:
- Early hyperventilation (respiratory alkalosis)
- Progression to hypoventilation and respiratory arrest as cerebral edema worsens
Milder or Partial Urea Cycle Defects (Late Onset)
- Ammonia accumulation may be triggered at any life stage by stressors (e.g., illness, fasting, surgery, peripartum period).
- Clinical features:
- Less severe hyperammonemia compared to neonatal presentation.
- Delayed initial presentation (months or years after birth).
- Common symptoms include:
- Loss of appetite, vomiting, lethargy
- Behavioral abnormalities, sleep disorders
- Delusions, hallucinations, psychosis
- EEG may show encephalopathic slow-wave patterns during episodes.
- MRI often reveals nonspecific brain atrophy.
Specific Disorders within the Urea Cycle
- ARG1 deficiency (Hyperargininemia):
- Neurological symptoms predominate, typically subtler presentation, though neonatal hyperammonemia can occur.
- Amino Acid Transporter Deficiencies:
- ORNT1 deficiency: May cause chronic liver dysfunction alongside hyperammonemia.
- Citrin deficiency: Typically presents later (adolescence/adulthood), may present in infancy with neonatal intrahepatic cholestasis, or in older children with failure to thrive.
Neurological Aspects of UCDs
- Ammonia-induced brain damage primarily involves cerebral edema due to increased glutamine levels.
- Acute hyperammonemia damage mimics hypoxic-ischemic or stroke patterns on MRI:
- Initially affects insular cortex and deep white matter.
- Prolonged cases affect parietal, occipital, and frontal regions.
- Early MRI may appear normal; clinical changes precede imaging abnormalities.
- Chronic hyperammonemia can disrupt:
- Ion gradients, neurotransmitter balance, mitochondrial function
- Alpha-ketoglutarate/glutamate/glutamine ratios
- Seizures are common, often subclinical, especially in neonates, correlating with rising glutamine even before peak ammonia levels [Wiwattanadittakul et al 2018].
Survival and Intellectual Outcomes
- Historically poor outcomes have significantly improved with rapid diagnosis and treatment
- Recent longitudinal NIH data demonstrate better intellectual outcomes with modern treatment protocols
- Additional factors, including nitric oxide pathway disruption, may affect intellectual outcomes regardless of hyperammonemia severity
- Asymptomatic female carriers of OTC deficiency demonstrate cognitive vulnerabilities during tasks requiring fine motor skills, executive function, and cognitive flexibility
Causes and Prevalence of Urea Cycle Disorders (UCDs)
Specific Urea Cycle Disorders
- N-acetylglutamate synthase deficiency (NAGS deficiency)
- Clinically resembles CPS1 deficiency, as CPS1 activity depends on N-acetylglutamate [Caldovic et al 2003].
- Carbamoylphosphate synthetase I deficiency (CPS1 deficiency)
- Most severe urea cycle disorder, characterized by rapid-onset neonatal hyperammonemia.
- Survivors remain at chronic risk for repeated hyperammonemic episodes.
- Ornithine transcarbamylase deficiency (OTC deficiency)
- Complete deficiency in males is as severe as CPS1 deficiency.
- About 15% of female carriers develop overt hyperammonemia requiring chronic treatment.
- Even asymptomatic carrier females may have subtle impairments in executive function.
- Citrullinemia type I (ASS1 deficiency)
- Severe hyperammonemia occurs but with slightly easier management due to partial incorporation of nitrogen into cycle intermediates.
- Argininosuccinic aciduria (ASL deficiency)
- Presents with rapid-onset neonatal hyperammonemia.
- Chronic hepatic enlargement and elevated transaminases may progress to liver fibrosis (etiology unclear).
- Characteristic trichorrhexis nodosa (fragile hair) typically responsive to arginine supplementation.
- Significant developmental disabilities common even in absence of prolonged coma [Summar 2001, Summar & Tuchman 2001, Nagamani et al 2012].
- Arginase deficiency (ARG1 deficiency, hyperargininemia)
- Typically slower onset; early severe presentations occasionally occur [Jain-Ghai et al 2011].
- Progressive spasticity, tremor, ataxia, choreoathetosis, and growth disturbances reported [Cederbaum et al 2004].
- Ornithine translocase deficiency (ORNT1 deficiency)
- Variable onset from infancy to adulthood.
- Clinical manifestations include chronic cognitive impairment, hyperammonemic crisis, or chronic liver dysfunction.
- Citrin deficiency
- Neonatal presentation as neonatal intrahepatic cholestasis (NICCD).
- Childhood presentation as failure to thrive and dyslipidemia (FTTDCD).
- Adult presentation as recurrent hyperammonemia with neuropsychiatric symptoms (Citrullinemia type II, CTLN2).
Prevalence of Urea Cycle Disorders
- Overall incidence is estimated at least 1:35,000 births; milder forms may increase actual prevalence.
Table 2. Estimated Incidence of Individual Urea Cycle Disorders
| Urea Cycle Disorder | Estimated Incidence |
|---|---|
| NAGS deficiency | <1:2,000,000 |
| CPS1 deficiency | 1:1,300,000 |
| OTC deficiency | 1:56,500 |
| ASS1 deficiency | 1:250,000 |
| ASL deficiency | 1:218,750 |
| ARG1 deficiency | 1:950,000 |
| Ornithine translocase deficiency | Unknown |
| Citrin deficiency | 1:100,000–1:230,000 in Japan1 |
- 1Incidence outside Japan unknown; East Asian carrier frequency ranges from 1:48 to 1:112, higher than expected based on incidence.
Evaluation Strategy to Identify the Specific Type and Genetic Cause of a Urea Cycle Disorder in a Proband
The diagnosis of a urea cycle disorder (UCD) in a symptomatic individual is based on clinical, biochemical, and molecular genetic assessments.
Family History
- Obtain a detailed three-generation family history with attention to relatives (especially children) presenting neurologic symptoms indicative of UCD.
- Review medical records for biochemical testing, molecular genetic results, or autopsy findings in affected relatives.
- An X-linked inheritance pattern strongly suggests OTC deficiency.
Physical Examination
- Generally nonspecific across UCD types.
- Distinctive findings suggestive of specific disorders:
- Trichorrhexis nodosa (fragile hair): indicative of ASL deficiency.
- Progressive lower-extremity spasticity: indicative of ARG1 deficiency.
Biochemical Testing
The following algorithm guides evaluation in newborns with hyperammonemia:
Steps in Evaluation of a Newborn with Hyperammonemia
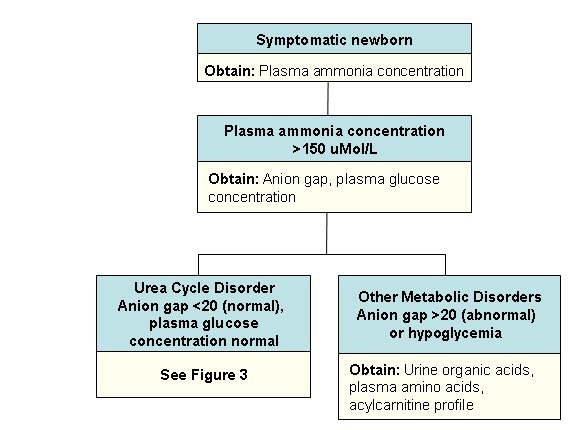
- Plasma ammonia concentration is typically the first abnormal laboratory finding:
- Ammonia concentration ≥150 μmol/L with a normal anion gap and normal glucose strongly suggests UCD [Summar & Tuchman 2001].
Diagnostic Tests for Urea Cycle Disorders
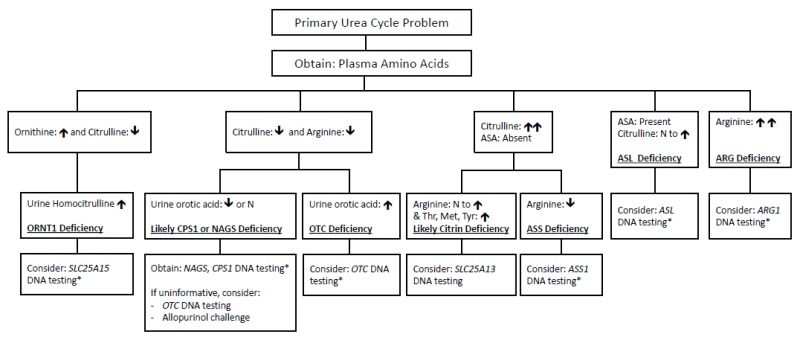
*Note: If DNA testing is inconclusive, enzymatic testing may be necessary.
- Quantitative plasma amino acid analysis provides a preliminary diagnosis:
- Plasma amino acid profiles differ in newborns compared to older individuals due to liver immaturity.
- Plasma citrulline concentration differentiates proximal versus distal enzyme defects:
- Absent or trace levels: neonatal-onset CPS1 deficiency, OTC deficiency, NAGS deficiency; also reduced in ORNT1 deficiency.
- Marked elevation: ASS1 deficiency.
- Moderate elevation (~2–5-fold): ASL deficiency, accompanied by elevated argininosuccinic acid (ASA) in plasma and urine. (Note: ASA is absent in unaffected individuals.)
- Moderately elevated citrulline with increased threonine/serine ratio: suggestive of citrin deficiency.
- Typically normal: ARG1 deficiency.
- Plasma arginine concentration:
- Markedly elevated: ARG1 deficiency.
- Reduced or occasionally normal: other UCDs (especially in partial defects).
- Plasma ornithine concentration:
- Elevated: ORNT1 deficiency (urine homocitrulline also elevated).
- Not elevated: OTC deficiency.
- Plasma glutamine, alanine, and asparagine concentrations:
- Often elevated due to their roles as nitrogen storage compounds.
- Urinary orotic acid:
- Distinguishes CPS1/NAGS deficiency (normal or low levels) from OTC deficiency (significantly elevated).
- Elevated also in ARG1 deficiency and ASS1 deficiency (citrullinemia type I).
- Urine amino acid analysis:
- Identifies elevated urine homocitrulline (ORNT1 deficiency).
- Useful for detecting argininosuccinic acid (ASA), which appears more prominently in urine compared to plasma, particularly when small peaks are challenging to detect on plasma analysis.
Molecular Genetic Testing for Urea Cycle Disorders (UCDs)
Molecular genetic testing is the primary method for definitive diagnostic confirmation for all eight UCDs. Although genetic testing has largely replaced enzymatic assays, enzyme activity testing remains valuable if genetic analysis is inconclusive.
Approaches to Molecular Genetic Testing
- Serial single-gene testing:
- Indicated when biochemical findings strongly suggest a specific gene mutation.
- Initial sequence analysis of the suspected gene.
- If sequencing is negative or inconclusive, follow-up gene-targeted deletion/duplication analysis is recommended:
- OTC deficiency: if hemizygous pathogenic variant is suspected but not initially identified.
- NAGS, CPS1, ASS1, ASL, ARG1, ORNT1, or citrin deficiencies: if fewer than two pathogenic variants are identified initially.
- Multigene panel testing:
- A comprehensive panel including the eight genes associated with UCDs can streamline diagnosis.
- Gene content and diagnostic sensitivity vary by laboratory and evolve over time.
- Panels may include unrelated genes; careful panel selection helps avoid incidental findings or variants of uncertain significance (VUS).
- Laboratories may offer customized gene panels or phenotype-focused exome analyses selected by clinicians.
- Typical methodologies: sequence analysis, deletion/duplication assays, and non-sequencing-based techniques.
- Comprehensive genomic testing:
- Exome sequencing or genome sequencing can be utilized if initial testing does not yield a definitive diagnosis.
- May identify atypical genetic causes or suggest alternative diagnoses with similar phenotypes.
Molecular Genetics of Urea Cycle Disorders
| Disease Name | Gene | Protein | Select OMIM Links |
|---|---|---|---|
| Carbamoylphosphate synthetase I deficiency | CPS1 | Carbamoyl-phosphate synthase | 608307, 237300 |
| Ornithine transcarbamylase deficiency | OTC | Ornithine carbamoyltransferase | 300461, 311250 |
| ASS1 deficiency (citrullinemia type I) | ASS1 | Argininosuccinate synthase | 603470, 215700 |
| ASL deficiency (argininosuccinic aciduria) | ASL | Argininosuccinate lyase | 608310, 207900 |
| Arginase deficiency | ARG1 | Arginase-1 | 608313, 207800 |
| NAGS deficiency | NAGS | N-acetylglutamate synthase | 608300, 237310 |
| Ornithine translocase (ORNT1) deficiency | SLC25A15 | Ornithine transporter (ORNT1) | 603861, 238970 |
| Citrin deficiency | SLC25A13 | Citrin | 603859, 605814, 603471 |
Enzyme Activity Testing (if Genetic Testing is Inconclusive)
- CPS1 deficiency, NAGS deficiency, OTC deficiency: hepatocyte assays.
- ASL deficiency, ASS1 deficiency, ORNT1 deficiency: fibroblast assays.
- ARG1 deficiency: erythrocyte assays.
Differential Diagnosis of Urea Cycle Disorders (UCDs)
Several conditions causing liver dysfunction can lead to hyperammonemia and clinical presentations similar to urea cycle disorders. These include hepatic diseases, medication-induced hyperammonemia, and various genetic disorders.
Diseases of the Liver and Biliary Tract
- Herpes simplex virus infection
- Vascular bypass of the liver
- Biliary atresia
- Acute liver failure
Medication-Induced Hyperammonemia
- Valproic acid
- Cyclophosphamide
- 5-pentanoic acid
Genetic Disorders in the Differential Diagnosis of UCDs
| Disorder | Gene(s) | MOI | Clinical Features Overlapping with UCD | Features Distinguishing from UCD |
|---|---|---|---|---|
| Propionic acidemia | PCCA, PCCB | AR | Hyperammonemia | Metabolic acidosis, hyperglycinemia, characteristic organic acids, acylcarnitine profile |
| Isolated methylmalonic acidemia | MUT, MMAA, MMAB, others | AR | Hyperammonemia | Metabolic acidosis, diagnostic organic acids, acylcarnitine profile |
| Isovaleric acidemia | IVD | AR | Hyperammonemia | Possible metabolic acidosis, characteristic organic acids, acylcarnitine profile |
| Carbonic anhydrase VA deficiency | CA5A | AR | Hyperammonemia | Elevated lactate or abnormal urine organic acids |
| Lysinuric protein intolerance | SLC7A7 | AR | Hyperammonemia | Elevated lysine, ornithine, arginine in urine |
| Fatty acid oxidation disorders (e.g., SCAD, MCAD, VLCAD) | Multiple genes | AR | Liver dysfunction | Characteristic elevations in diagnostic acylcarnitines |
| Hyperinsulinism-hyperammonemia syndrome | GLUD1 | AD | Hyperammonemia | Hypoglycemia, hyperinsulinism |
| OAT deficiency (neonatal) | OAT | AR | Hyperammonemia | Ornithine low in neonates; older individuals show elevated ornithine without hyperammonemia |
| Tyrosinemia type I | FAH | AR | Liver dysfunction | Diagnostic amino acids, presence of succinylacetone |
| Classic galactosemia | GALT | AR | Liver dysfunction | Elevated galactose-1-phosphate, reduced galactose-1-phosphate uridyltransferase enzyme activity |
| Mitochondrial disorders | Multiple genes | AR, mt, XL | Liver dysfunction, occasionally reduced citrulline | Increased plasma alanine, elevated plasma lactate |
Abbreviations:
- AD = Autosomal dominant
- AR = Autosomal recessive
- MOI = Mode of inheritance
- MCAD = Medium-chain acyl-CoA dehydrogenase deficiency
- SCAD = Short-chain acyl-CoA dehydrogenase deficiency
- VLCAD = Very long-chain acyl-CoA dehydrogenase deficiency
- mt = Mitochondrial inheritance
- XL = X-linked
- OAT = Ornithine aminotransferase
Genetic Risk Assessment in Family Members of Individuals with Urea Cycle Disorders (UCDs)
Genetic counseling provides families information on inheritance patterns, genetic risks, and implications of genetic disorders, aiding informed medical and personal decisions. The following information outlines genetic risk assessment in urea cycle disorders (UCDs). For personalized genetic counseling, professional genetics consultation is recommended.
Modes of Inheritance
- OTC deficiency: X-linked inheritance.
- All other UCDs (NAGS, CPS1, ASS1, ASL, ARG1, ORNT1, citrin deficiencies): Autosomal recessive inheritance.
X-Linked Inheritance – OTC Deficiency
Risks to Family Members
- Parents of a Male Proband:
- Father:
- Unaffected and not hemizygous; no further testing required.
- Mother:
- Obligate heterozygote (carrier) if multiple affected family members exist.
- If a single affected male exists (simplex case), the mother may be a carrier or the affected male may have a de novo pathogenic variant (approximately 26% of cases) [Rüegger et al 2014].
- If a mother has multiple affected children but no detectable OTC variant in leukocyte DNA, germline mosaicism is likely (reported, but frequency unknown; general risk estimated at 3%-4%) [Bowling et al 1999].
- Father:
- Parents of a Female Proband:
- A female proband can inherit the OTC pathogenic variant from either parent, or the variant may occur de novo.
- Molecular testing of parents and detailed family history review help clarify inheritance.
- Siblings of a Proband (Risk depends on parental genetic status):
- If the mother is a carrier:
- 50% chance per pregnancy of inheriting the pathogenic variant:
- Affected males develop disease.
- Females may or may not exhibit clinical symptoms.
- 50% chance per pregnancy of inheriting the pathogenic variant:
- If the father is affected (for female probands):
- All daughters inherit the pathogenic variant; sons do not inherit the variant.
- In simplex cases with no detectable maternal variant, sibling risk is low but higher than the general population due to potential maternal germline mosaicism.
- If the mother is a carrier:
Note: Additional details are available under "OTC Deficiency: Heterozygous Females, Evaluation of Relatives at Risk, and Risk to Family Members."
Autosomal Recessive Inheritance (NAGS, CPS1, ASS1, ASL, ARG1, ORNT1, Citrin deficiencies)
Risks to Family Members
- Parents of a Proband:
- Both parents are obligate carriers (heterozygotes).
- Carriers remain asymptomatic and have no risk of developing disease.
- Siblings of a Proband:
- Each sibling has:
- 25% chance of being affected.
- 50% chance of being an asymptomatic carrier.
- 25% chance of being unaffected and not a carrier.
- Carrier siblings remain asymptomatic.
- Each sibling has:
- Offspring of a Proband:
- All offspring are obligate heterozygotes (carriers).
- Carriers remain asymptomatic.
Carrier Detection
- Carrier testing requires prior identification of specific pathogenic variants in the family.
Further reading
Ah Mew N, Simpson KL, Gropman AL, et al. Urea Cycle Disorders Overview. 2003 Apr 29 [Updated 2017 Jun 22]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1217/
