Introduction
-
Forms of Neurofibromatosis:
- Neurofibromatosis Type 1 (NF1): Previously known as von Recklinghausen disease, the most common type.
- NF2-related Schwannomatosis (NF2): Formerly neurofibromatosis type 2.
- Schwannomatoses: Related to genetic variants other than NF2.
-
Hallmarks of NF1:
- Multiple café-au-lait macules.
- Neurofibromas.
- Segmental NF1: Clinical features limited to one body area due to somatic mosaicism of a pathogenic variant in the NF1 gene.
Epidemiology
-
Incidence:
- NF1: Autosomal dominant genetic disorder with an incidence of 1:2600 to 1:3000 individuals.
- Approximately 50% of cases are familial (inherited).
- De novo mutations occur primarily in paternally derived chromosomes.
- Segmental NF1: Prevalence estimated at 1:36,000 to 1:40,000.
-
Prevalence Study:
- Population-based study in Finland: Overall prevalence of NF1 approximately 1:4000.
- Prevalence decreases with age.
- Hazard ratio of death among individuals with NF1: 3.10.
Pathogenesis
-
Genetic Basis:
- Pathogenic variants in the NF1 gene located at chromosome 17q11.2.
- Protein product: Neurofibromin, a GTPase-activating protein (GAP) family member.
- Involvement in signaling pathways: SCF/c-kit signaling, mTOR, and MAPK pathways.
-
Clinical Findings:
- Pathogenic variants result in loss of production or reduced function of neurofibromin.
- Complete penetrance but highly variable expression.
- Somatic mutation or loss of heterozygosity at the NF1 locus leads to complete loss of neurofibromin expression in NF1 lesions such as pseudoarthrosis and neurofibromas.
- NF1 functions as a tumor suppressor gene.
- Haploinsufficiency may account for some phenotype aspects, such as neurocognitive problems.
-
Segmental NF1:
- Caused by somatic mosaicism due to a postzygotic mutation in the NF1 gene.
- Some cells have two fully functional NF1 genes; other cells contain a pathogenic variant in one copy of the NF1 gene.
- Individuals with segmental NF1 do not have an affected parent.
- Adults with localized NF1 and mosaicism in somatic and gonadal tissues can transmit the mutation to offspring, resulting in non-segmental manifestations.
- Rare cases of germline mosaicism without apparent somatic features have been described.
Clinical Manifestations
-
Features of Neurofibromatosis Type 1 (NF1) by Age:
- Typical Order of Appearance:
- Café-au-lait macules
- Axillary and/or inguinal freckling
- Lisch nodules (iris hamartomas)
- Neurofibromas
- Early Manifestations:
- Osseous dysplasias: Usually appear during the first year after birth.
- Symptomatic optic pathway glioma (OPG): Typically present by three years of age.
- Later Manifestations:
- Other tumors and neurologic complications: Begin after the first year of life.
- Hypertension: May occur in childhood.
- Malignant transformation of tumors: More common in adolescence and adulthood.
- Typical Order of Appearance:
-
Café-au-lait Macules:
- Description: Flat, uniformly hyperpigmented macules.
- Onset: Appear during the first year after birth, increase in number during early childhood.
- Prevalence: Up to 15% of the general population has one to three café-au-lait macules.
- Significance: Presence of six or more macules is highly suggestive of NF1.
- Study Findings: Younger age at presentation (≤29 months) and six or more macules indicate high risk of NF1 (80.4%).
- Visualization: Wood's lamp can help visualize macules in older individuals when they are not readily discernible.
-
Freckling:
- Description: Freckling in axillary or inguinal regions (Crowe sign), smaller than café-au-lait macules, occur in clusters.
- Onset: Usually appears by age three to five years, typically first in the inguinal region.
- Location: Occurs mostly in skin folds, especially axillary and inguinal areas; may also appear in other intertriginous areas like neckline or inframammary areas.
-
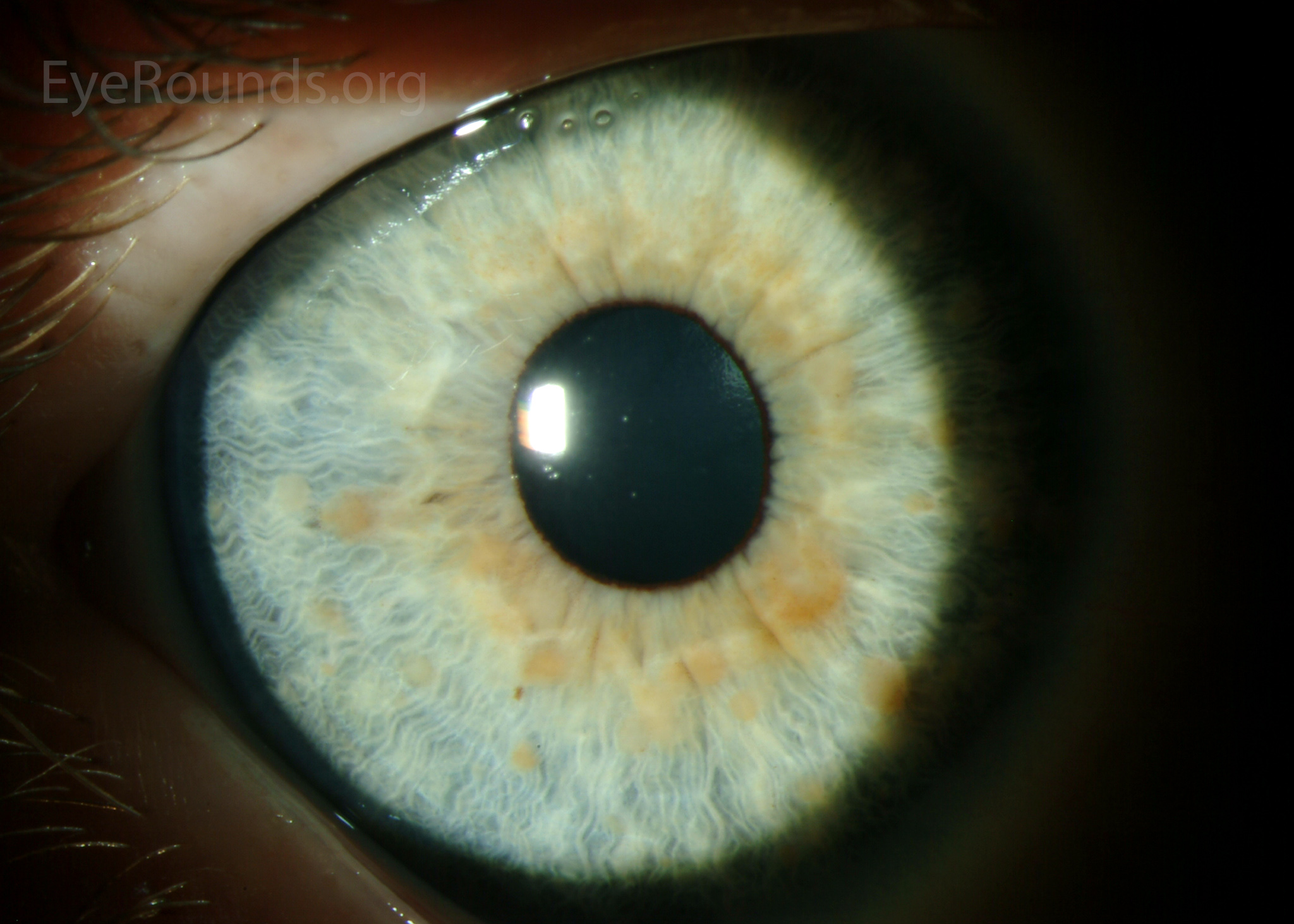
Ocular Features:
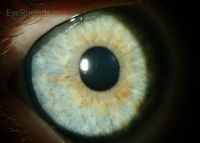
- Lisch Nodules:
- Description: Raised, tan-colored hamartomas of the iris.
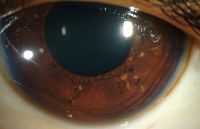 Lisch nodules LRG
Lisch nodules LRG - Prevalence: Detected in fewer than 10% of children under six years of age but seen in over 90% of adults.
- Detection: Visible with a direct ophthalmoscope or slit lamp.
- Description: Raised, tan-colored hamartomas of the iris.
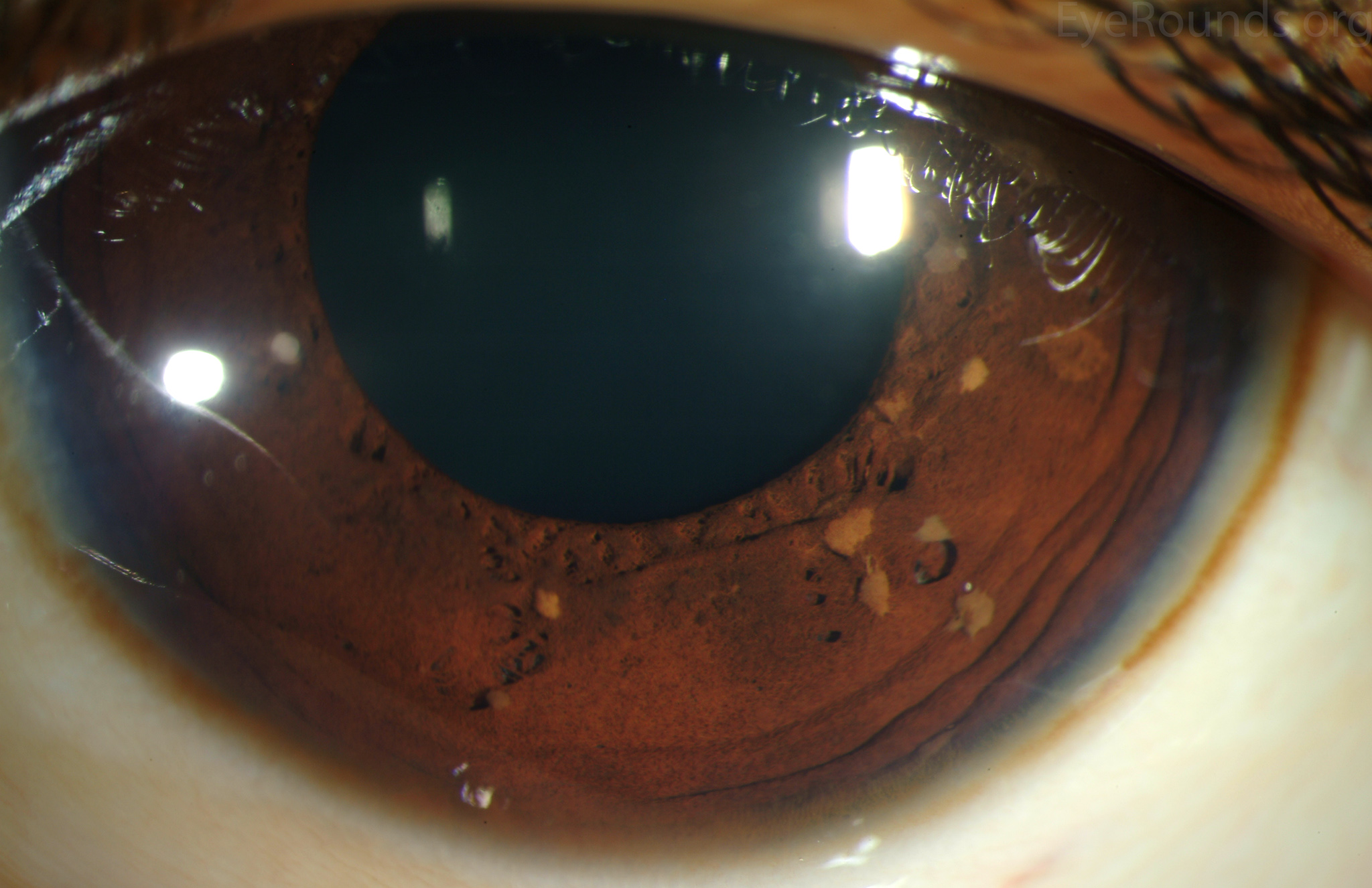
- Choroidal Abnormalities:
- Description: Aggregations of melanocytes.
- Detection: Visualized with near-infrared reflectance imaging and optical coherence tomography.
- Prevalence: More common than Lisch nodules (64% vs. 41%) in children aged 3 to 18 years.
- Lisch Nodules:
Tumors
-
General Overview:
- Increased Frequency: Persons with NF1 develop both benign and malignant tumors more frequently throughout life.
 Lisch nodules
Lisch nodules - Common Types:
- Benign Tumors: Neurofibromas are the most common.
- Intracranial Neoplasms: OPGs and other gliomas.
- Non-CNS Malignancy: Malignant peripheral nerve sheath tumors (MPNSTs).
- Risk of Malignancy: Higher compared to the general population, driven by MPNST.
- Cancer Diagnosis: Occurs at a younger age in NF1 individuals.
- Increased Frequency: Persons with NF1 develop both benign and malignant tumors more frequently throughout life.
-
Peripheral Neurofibromas:
- Description: Benign peripheral nerve sheath tumors composed of Schwann cells, fibroblasts, perineurial cells, mast cells, macrophages, and T cells.
- Cellular Basis: Loss of both NF1 alleles occurs in Schwann cells.
- Growth Patterns:
- Focal growths or longitudinally along a nerve (plexiform neurofibromas).
- Locations: Skin (cutaneous neurofibromas), along peripheral nerves under the skin or deeper inside the body, and along nerve roots adjacent to the spine.
- Pregnancy Influence: Number and size of neurofibromas may increase during pregnancy.
-
Growth Trends in Adulthood:
- Study Findings: 63% of internal tumors shrank by ≥20% without treatment over 10.4 years; only 17% enlarged by ≥20%.
-
Cutaneous Neurofibromas:
- Description: Soft, fleshy, sessile or pedunculated tumors.
- Onset: Usually appear before or during adolescence.
- Characteristics: Increase in size and number with age, highest density over the trunk.
- Cosmetic Impact: Major issue in adults, associated with pruritus during accelerated growth.
-
Plexiform Neurofibromas:
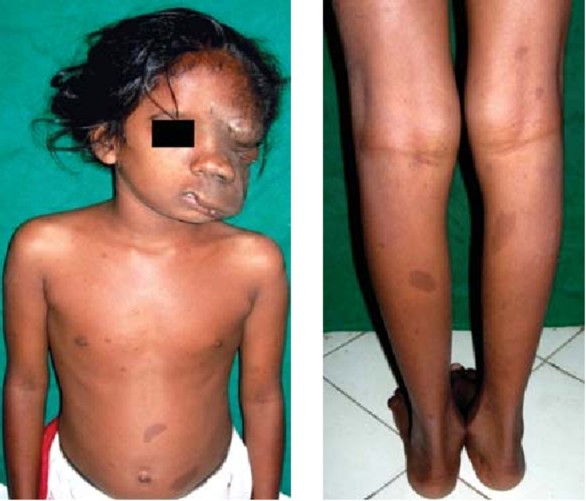 Plexiform Neurofibroma & Café au lait macules (Source: Misra S (2012))
Plexiform Neurofibroma & Café au lait macules (Source: Misra S (2012))- Location: May be superficial, associated with overgrowth of skin and soft tissues, or deep inside the body.
- Growth: Tend to grow most rapidly during childhood.
- Prevalence: Found in approximately 50% of patients with NF1.
- Impact: Major cause of morbidity and disfigurement, associated with increased mortality.
- Complications: Can compress the airway or spinal cord, potentially transform into MPNSTs.
- Orbital Plexiform Neurofibromas: Associated with sphenoid wing dysplasia, leading to enophthalmos or exophthalmos.
-
Nodular Neurofibromas:
- Description: Discrete lesions that appear as firm, rubbery masses under the skin or deeper inside the body.
- Characteristics: Can compress surrounding structures or cause pain, do not invade surrounding tissues like plexiform neurofibromas.
- Atypical Neurofibromas: May exhibit nuclear atypia, some mitotic activity, and dense cellularity. Identified as "atypical neurofibroma" or "atypical neurofibromatous neoplasms of uncertain biologic potential" (ANNUBP).
- Genetic Basis: Homozygous deletion of cyclin-dependent kinase inhibitor 2A/B (CDKN2A/B) along with loss of both NF1 alleles.
Optic Pathway Gliomas (OPGs)
-
Prevalence:
- Occur in approximately 15% of children younger than six years of age with NF1.
- Rarely occur as new tumors in older children and adults.
-
Characteristics:
- Typically low-grade pilocytic astrocytomas.
- Can arise anywhere along the anterior visual pathway to the optic radiations.
- Involve optic nerves, chiasm, and postchiasmal optic tracts.
-
MRI Study Findings:
- Prevalence of optic pathway tumors in a study of 562 adults and children with NF1 was 9.3%.
- Distribution in 52 persons with optic glioma:
- 29 had intraorbital tumors.
- 32 had prechiasmatic tumors.
- 17 had involvement of the optic chiasm.
- 19 had extension to the optic radiations.
- 29 had involvement of two or more regions.
- Visual decline was found in 17 of 52 persons, with 7 requiring treatment.
-
Vision and Symptoms:
- Many children with NF1 and OPGs have normal vision.
- A minority become symptomatic with progressive vision loss.
- Risk Factors for Visual Loss:
- Age less than two years.
- Female sex.
- Tumor involvement of the postchiasmal optic pathway.
- Symptoms and Signs:
- Decreased visual acuity or color vision.
- Abnormal pupillary function.
- Proptosis.
- Optic nerve atrophy.
- Assessment: Optical coherence tomography can assess retinal nerve fiber thickness to follow visual function over time.
-
Premature or Delayed Puberty due to OPGs:
- Tumors involving the optic chiasm may cause either premature or delayed puberty due to hypothalamic involvement.
- Importance of Early Detection:
- Precocious puberty may indicate the presence of a clinically significant OPG.
- Early treatment can minimize complications of accelerated linear bone growth and premature development of secondary sexual characteristics.
- One of the earliest signs of precocious puberty is accelerated linear growth.
- Growth Charts: Standards for children with NF1 are available to monitor growth accurately.
Other Central Nervous System Neoplasms
-
Increased Risk:
- Individuals with NF1 are at an increased risk for developing various CNS neoplasms, in addition to OPGs.
- Common Types:
- Low-grade astrocytomas
- Brainstem gliomas
- High-grade gliomas
-
Clinical Presentation:
- Most frequent presentation: Increased intracranial pressure.
- Many lesions are asymptomatic and are discovered incidentally during brain imaging.
- Symptomatic lesions are more likely to progress and require treatment.
-
Brainstem Gliomas:
- Children with NF1:
- May be asymptomatic and not require therapy.
- Symptomatic brainstem gliomas in children are more likely to be higher-grade tumors (WHO grade 3 or 4) requiring treatment.
- Adults with NF1:
- Suspected gliomas (brainstem or otherwise) should undergo biopsy whenever feasible to confirm an integrated molecular diagnosis and guide therapy.
- Clinical behavior of gliomas in adults with NF1 is often aggressive, even for low-grade tumors.
- Children with NF1:
Soft Tissue Sarcomas
-
Increased Risk:
- Persons with NF1 have a higher risk of developing soft tissue sarcomas, such as MPNSTs, rhabdomyosarcoma (RMS), and gastrointestinal stromal tumors (GISTs).
-
Malignant Peripheral Nerve Sheath Tumors (MPNSTs):
- Previous Name: Neurofibrosarcomas.
- Origin: Typically arise within preexisting plexiform or atypical neurofibromas.
- Clinical Signs: Significant and constant pain, change in consistency from soft to hard, and/or rapid growth of a nodule within an existing plexiform neurofibroma.
- Imaging:
- MRI: Large size, depth below the fascial layer, and necrosis suggest malignant change.
- PET Imaging: 18-fluorodeoxyglucose PET helps distinguish MPNST from benign neurofibromas.
- Diffusion-Weighted MRI: Useful in differentiating benign neurofibromas, atypical neurofibromas, and MPNSTs.
- Lifetime Risk:
- Varies from 8 to 13 percent, with a higher risk at older ages.
- Risk by age: 8.5% by age 30, 12.3% by age 50, and 15.8% by age 85.
- Among children with MPNSTs, 20 to 50 percent have NF1.
- Tumor Characteristics: NF1-associated tumors tend to be larger at diagnosis.
-
Rhabdomyosarcoma (RMS):
- Frequency: More common in persons with NF1.
- Presentation: Often arises at an early age, typically in a genitourinary site.
- Histology: Usually of the embryonal subtype.
-
Gastrointestinal Stromal Tumors (GISTs):
- Risk: Increased in persons with NF1.
- Prevalence: 3.7% in a cohort of 108 adults with NF1.
- Characteristics:
- Frequently occur in the small intestine (over 70%).
- Often multiple.
- Different molecular pathology compared to sporadic GISTs.
-
Glomus Tumors:
- Location: Arise in the tips of fingers and toes under the nail bed.
- Symptoms: Present with pain, tenderness, and sensitivity to cold.
- Treatment: Pain relieved by surgical removal of the tumor.
Other Tumors
-
Juvenile Myelomonocytic Leukemia and Pheochromocytoma:
- Increased Risk: Higher incidence in individuals with NF1.
- Other Genetic Etiologies: More common causes for these malignancies outside NF1.
-
Breast Cancer:
- Increased Risk: Higher in females with NF1, particularly those under 50 years of age.
- Characteristics:
- Often estrogen receptor negative.
- HER2 positive.
- Associated with younger age at diagnosis and less favorable prognosis compared to non-NF1-associated breast cancers.
-
Other Malignancies:
- Higher Rates: Neuroendocrine tumors, melanoma, ovarian cancer, and Hodgkin lymphoma.
- Uncertainty: Whether other common malignancies occur at higher rates is not fully established.
Bone Abnormalities
-
General Overview:
- Bone abnormalities in NF1 include pseudoarthrosis, bone dysplasia, short stature, scoliosis, nonossifying fibromas, sphenoid dysplasia, and osteoporosis.
-
Long Bone Dysplasia and Pseudoarthrosis:
- Prevalence: Affects approximately 5% of infants or young children with NF1.
- Common Presentation: Anterolateral bowing of the tibia, progressing to narrowing of the medullary canal, cortical thickening, and fracture.
- Diagnosis: Often overlooked until pathologic fractures occur with weight bearing or walking attempts.
- Pseudoarthrosis: False joint formation due to nonunion of bone fragments at a long bone fracture site, severely compromising limb function.
- NF1 as a Cause: Most common cause of long bone pseudoarthrosis, accounting for 50-80% of cases.
-
Other Bone Lesions:
- Vertebral Defects: Scalloping caused by dural ectasia.
- Nonossifying Fibromas: Within long bones.
- Sphenoid Wing Dysplasia: May present as facial asymmetry.
- Lambdoidal Suture Defect: Rare, usually on the left side of the head.
-
Growth:
- Short Stature: Common in children with NF1.
- Database Findings: 13% of 569 White North American patients with NF1 had a height ≥2 standard deviations below the population mean.
- Puberty: Decreased height velocity during puberty, more prominent in males.
- Growth Curves: Available for children with NF1 to determine if short stature is consistent with NF1 or suggests another cause.
- Birth Weight: Increased in infants with NF1, though maternal NF1 is associated with decreased birth weight.
-
Scoliosis:
- Prevalence: Occurs in 10-25% of persons with NF1.
- Onset: Frequently becomes apparent at 6-10 years of age or in early adolescence.
- Characteristics: Most commonly affects the thoracic spine, tends to be sharply angulated and dystrophic.
- Associated Conditions: Sometimes associated with paraspinal plexiform neurofibroma.
-
Osteoporosis:
- Bone Density: Persons with NF1 have lower bone density compared to age-matched general population controls.
- Severity: Can range from osteopenia to osteoporosis.
- Etiology: Unknown, largely independent of vitamin D-related dysregulation.
- Fracture Risk: Increased risk of fractures in both children and adults over 40 years of age with NF1, compared to population controls of similar age and sex distribution.
Neurologic Abnormalities
-
General Overview:
- Neurologic disorders in NF1 include cognitive deficits, learning disabilities, headaches, seizures, gross and fine motor developmental delays, macrocephaly, and symptoms from dural ectasia along the spine causing nerve root compression and pain.
-
Cognitive Deficits and Learning Disabilities:
- Frequency: Higher in children with NF1.
- Study Findings: 81% of children with NF1 have moderate to severe impairment in one or more cognitive domains.
- IQ Scores: 5 to 10 points lower compared to the general population or unaffected sibling controls.
- Incidence of Intellectual Disability: 4 to 8%, slightly higher than the general population (2 to 3%).
- Associated Conditions: Increased frequency of autism spectrum disorder.
- Learning Disabilities: Up to 65% of children with NF1, poor performance on tasks involving nonverbal learning, visuospatial function, language-based learning, and impaired social skills.
- Attention Deficit Hyperactivity Disorder (ADHD): Occurs in 30 to 40% of children with NF1.
- Executive Function Impairment: Issues with planning and problem-solving not directly related to inattention.
- Speech and Auditory Processing: Problems with speech articulation, hypernasal voice, muscle weakness, auditory processing deficits, and impaired speech discrimination.
- Motor Development: Fine and gross motor developmental problems seen early, with the gap widening over time compared to peers.
- Sleep Disorders: Also common in children with NF1.
-
Seizures:
- Prevalence: Twice as common in NF1 patients (4 to 6%).
- Types: Can be of any type and begin at any age, usually not attributable to a brain mass lesion.
- Imaging: New seizures should prompt repeat neuroimaging, even if previous imaging was normal.
-
Macrocephaly:
- Head Size: Generally larger in persons with NF1.
- Presentation: Relative or absolute macrocephaly due to increased brain volume.
- Complications: Rarely, hydrocephalus from aqueductal stenosis and Chiari malformation.
-
Peripheral Neuropathy:
- Prevalence: Less common in NF1 than in neurofibromatosis type 2 (NF2).
- Nerve Compression: Reported in up to 4% of patients, spinal root compression in up to 3%.
- Comparison to NF2: Peripheral neuropathy symptoms occur in almost 50% of NF2 individuals.
- Severity: Can be severe, associated with morbidity, and may indicate a more severe NF1 phenotype.
- Forms: Polyneuropathy can occur without compressive lesions, may take the form of small fiber neuropathy.
Congenital Heart Disease (CHD)
- Increased Incidence: Higher incidence in NF1.
- Study Findings:
- 12.6% (62 out of 493 genotyped NF1 patients) had some form of CHD.
- Common Types:
- Pulmonic stenosis (21 persons)
- Mitral valve anomalies (20 persons)
- Septal defects (10 persons)
- Less Common Types:
- Tetralogy of Fallot
- Thickening of the ventricular wall
- Associations: Those with CHD were more likely to have Noonan syndrome-like facial features and either whole gene deletions or nontruncating NF1 gene mutations.
Hypertension
- Prevalence: Frequent finding in adults with NF1, may develop during childhood.
- Types:
- Primary (Essential) Hypertension: Most cases.
- Renovascular Hypertension: More frequent due to vascular lesions.
- Evaluation: Renal artery stenosis evaluation recommended for children with NF1 and hypertension.
- Renovascular lesions can be present in normotensive patients, frequency of hypertension development unknown.
- Renal Dysfunction: Increased frequency of renal tubular and glomerular dysfunction compared to healthy controls.
- Pheochromocytoma: Less common cause, important to consider in hypertensive NF1 patients due to potential morbidity.
- Prevalence: 2.9% in a retrospective chart review of 1415 NF1 individuals.
- Age of Diagnosis: Median age of 41 years, with some cases in individuals under 21 years old.
- Screening Recommendations: Plasma fractionated metanephrines or 24-hour urine fractionated metanephrines and catecholamines every three years in asymptomatic patients, though guidelines suggest screening only in the presence of symptoms.
Other Manifestations
- Gastrointestinal Issues: Increased risk of constipation and irritable bowel syndrome.
- Cardiovascular and Respiratory Issues:
- Rarely, cardiovascular complaints or airway compromise due to mediastinal neurofibromas or MPNST metastases to the heart and lung.
- Pulmonary Conditions:
- Pulmonary hypertension
- Pulmonary artery stenosis
- Interstitial lung disease
- Bullous lung disease
- More common in adults, but radiographic signs can be seen in children.
- Vascular Lesions:
- May cause stenosis of major vessels, including the internal carotid, resulting in moyamoya disease.
- Rare instances of arterial dissection, sometimes leading to life-threatening hemorrhage.
Quality of Life
- Impact: Persons with NF1 report decreased quality of life due to diverse manifestations, neurocognitive problems, risk of disfigurement, and even death.
- Psychosocial Issues:
- Problems with self-image, anxiety, and chronic pain in both adults and children.
- Increased frequency of psychiatric problems.
- Decreased educational attainment.
- Overall impairment of quality of life.
Segmental NF1
- Cause: Due to mosaicism for an NF1 gene mutation.
- Presentation:
- Similarity to Generalized NF1: Similar age of appearance for specific features, with a few exceptions.
- Pigmentary Features and Plexiform Neurofibromas: Tend to present in children.
- Cutaneous Neurofibromas: Develop in adults.
- Lisch Nodules: May be present in one or both eyes.
- Affected Area:
- Typically limited to one side of the body.
- Involvement can range from a narrow strip to one-half of the body.
- Some individuals with mosaicism may have generalized manifestations.
- Complications:
- More serious complications of NF1, such as OPGs, pseudoarthrosis, plexiform neurofibromas, and learning difficulties, are uncommon.
- In a series of 124 patients, these serious complications occurred in 5.6% of those with segmental NF1.
Neuroimaging Findings
-
General Overview: Brain neuroimaging abnormalities are frequently detected in persons with NF1, including NF-associated bright spots and increased brain volume.
-
Focal Areas of High Signal Intensity (FASI):
- Terminology: Also called "unidentified bright objects" or neurofibromatosis bright spots. The term "unidentified bright objects" is discouraged to avoid distress.
- Prevalence: Found in 40 to 95 percent of pediatric patients with NF1.
- Common Locations: Basal ganglia, cerebellum, brainstem, and subcortical white matter.
- Characteristics:
- Represent increased fluid within myelin associated with dysplastic glial proliferation (vacuolar myelinopathy).
- Usually less than 15 mm in diameter, may reach up to 25 mm.
- Contrast enhancement is atypical.
- Lesions are dynamic; they may appear or increase in size/number in childhood and spontaneously regress during adolescence or puberty.
- Not malignant or premalignant, and not associated with focal neurologic deficits.
- Association with Brain Tumors: One retrospective study found that 28 percent of children with FASI developed a brain tumor requiring treatment, most often in the basal ganglia or cerebellum.
- Cognitive Function:
- Conflicting data on the relationship between FASI and cognitive function.
- Some studies report lower IQ and language scores, impaired visuomotor integration and coordination in children with FASI.
- Other studies find no association with intellectual impairment.
-
Increased Brain Volume (Megalencephaly):
- Prevalence: Brain volumes are increased in individuals with NF1.
- Associated Conditions: Linked with macrocephaly and cognitive deficits and learning disabilities.
-
Cerebrovascular Dysplasia:
- Prevalence: Noted in 2 to 6 percent of children with NF1 who underwent neuroimaging.
- Common Conditions:
- Moyamoya disease
- Intracranial aneurysm
- Clinical Presentation: Patients may be asymptomatic despite angiographic progression, but others may require surgery for revascularization.
Diagnosis
-
Revised Diagnostic Criteria for Neurofibromatosis Type 1 (NF1):
- The diagnosis is based on characteristic clinical features. Genetic testing can aid in establishing the diagnosis, especially in children who do not meet the criteria or only show café-au-lait macules and skin fold freckling.
-
Evaluation by Multidisciplinary Team:
- Should include pediatric neurologists, geneticists, and ophthalmologists.
- The team should:
- Examine for diagnostic criteria and treatable complications.
- Provide anticipatory guidance.
- Refer to specialists as needed.
-
Initial Screening Evaluation:
- Confirm the diagnosis by identifying clinical features of NF1.
- Obtain history regarding symptoms like pain, visual complaints, weakness, neurologic deficits, headaches, and seizures.
- Review developmental history and school progress.
- Focus physical examination on skin, skeletal, and neurologic systems.
- Perform ophthalmologic evaluation to identify Lisch nodules, choroidal abnormalities, and early signs of optic pathway glioma (OPG).
Diagnostic Criteria
- NIH Consensus Criteria (updated in 1997 and 2021):
- In an individual without an NF1-affected parent, the diagnosis is established if at least two of the following criteria are met:
- Six or more café-au-lait macules >5 mm in diameter in prepubertal and >15 mm in diameter in postpubertal individuals (measured in ordinary room light, not with a Wood's lamp).
- Two or more neurofibromas of any type or one plexiform neurofibroma.
- Freckling in the axillary or inguinal region.
- Optic pathway glioma (OPG).
- Two or more Lisch nodules (iris hamartomas) or two or more choroidal abnormalities.
- A distinctive bony lesion, such as sphenoid dysplasia, anterolateral bowing of the tibia, or long bone pseudoarthrosis.
- A heterozygous pathogenic NF1 variant with a variant allele fraction of 50 percent in apparently normal tissue (e.g., white cells).
- A child with an NF1-affected parent can be diagnosed if one or more of these criteria are met.
- In an individual without an NF1-affected parent, the diagnosis is established if at least two of the following criteria are met:
Specificity and Sensitivity
- The diagnostic criteria are highly specific and sensitive except in the youngest children.
- 97 percent of affected persons meet the original NIH criteria by eight years of age, and the remainder by 20 years of age.
- In a study of 1893 persons with NF1 younger than 21 years, approximately 46 percent of sporadic NF1 cases did not meet the criteria by one year of age.
- Young children with only one clinical manifestation and no family history should be monitored for the appearance of other manifestations.
- Genetic testing can be considered for making a molecular diagnosis.
Genetic Testing
-
Purpose:
- Confirm diagnosis in questionable cases.
- Direct screening of family members (targeted testing for the mutation identified in the proband).
- Required for prenatal or preimplantation diagnosis.
-
Mutation Impact:
- A positive NF1 mutation test does not predict the severity or complications of the disorder, with some exceptions.
-
Testing Scope:
- Sequencing of the entire coding region.
- Tests for deletions or rearrangements of the entire gene or portions of it.
- Examination for exonic or intronic variants that disrupt splicing.
-
Availability and Accuracy:
- Molecular testing is clinically available.
- Finds the causative pathogenic variant in approximately 95% of patients with a clinical diagnosis of NF1.
- A negative test does not completely exclude the diagnosis and may indicate mosaicism or a different disorder.
-
Pathogenic Variants:
- Thousands of distinct pathogenic NF1 variants have been identified.
- Few genotype-phenotype correlations, especially in small mutations (<20 base pairs).
- Specific mutations with known correlations:
- c.2970-2972 delAAT (p.M992del): Very mild phenotype.
- Missense variants in codons 844 to 848: Severe phenotype with multiple complications.
- p.Met1149, p.Arg1276, or p.Lys1423: Noonan syndrome-like phenotype, with some associated with congenital heart defects and high burden of spinal neurofibromas.
-
Large Deletions:
- 1 to 5% of NF1 patients have large deletions encompassing 1.4 to 1.5 Mb of DNA.
- Associated with higher incidence of intellectual disability, developmental delay, dysmorphic facial features, connective tissue abnormalities, and earlier appearance of cutaneous neurofibromas.
- Increased risk of malignant peripheral nerve sheath tumors (MPNSTs).
-
Differential Diagnosis:
- Conditions such as Legius syndrome, constitutional mismatch repair-deficiency (CMMR-D) syndrome, and Noonan syndrome can manifest similar features (café-au-lait macules and axillary freckling).
- Genetic testing helps in the diagnosis for patients who meet only two NIH criteria or only one criterion.
Screening Family Members
- Family History:
- Obtain a detailed family history to detect possible NF1 symptoms when diagnosed in a child.
- Examine biologic parents and siblings for characteristic signs.
- Include a complete skin examination and slit lamp examination of the eyes.
Prenatal Testing
- Methods:
- Amniocentesis or Chorionic Villus Sampling: Obtain sample for fetal genotyping if the precise mutation of an affected family member is known.
- Preimplantation Genetic Testing (PGT): Identify embryos that do not carry a known familial NF1 mutation.
Differential Diagnosis
The differential diagnosis of NF1 includes Legius syndrome, constitutional mismatch repair-deficiency (CMMR-D) syndrome, NF2-related schwannomatosis (NF2), and Noonan syndrome. Genetic testing can help distinguish NF1 from these other syndromes. The table below provides a differential diagnosis of café-au-lait macules.
Legius Syndrome
- Clinical Features: Includes a subset of NF1 features (multiple café-au-lait macules, axillary freckling, macrocephaly) but lacks neurofibromas and CNS tumors.
- Inheritance: Autosomal dominant disorder resulting from germline loss-of-function variants in SPRED1.
- Mechanism: SPRED1 is a negative regulator of Ras-Raf kinase interaction and MAPK signaling. Complete SPRED1 inactivation is needed to generate a café-au-lait spot.
Constitutional Mismatch Repair-Deficiency (CMMR-D) Syndrome
- Inheritance: Autosomal recessive disorder caused by deleterious variants in both copies of one of the four mismatch repair genes (MLH1, MSH2, MSH6, PMS2).
- Clinical Manifestations:
- Shares café-au-lait macules, axillary freckling, and Lisch nodules with NF1.
- Main difference: Types of malignancies seen.
- Hematologic malignancies in infancy to early childhood.
- Brain tumors (primarily glioblastoma) in midchildhood.
- Colorectal cancer in adolescence to young adulthood.
- Less common tumors: RMS, OPG.
- Lynch Syndrome: Caused by heterozygous pathogenic variants in one of the MMR genes.
- Testing Guidelines: Recommended for those presenting with one diagnostic feature of NF1 (usually café-au-lait spots), negative for NF1 and SPRED1, with specific family or personal history.
NF2-related Schwannomatosis
- Genes: NF2 and NF1 are caused by pathogenic variants in different genes.
- Key Differences:
- Café-au-lait macules can be seen but are less frequent in NF2.
- Lisch nodules are not seen in NF2.
- Schwannomas in NF2 do not undergo malignant transformation into MPNST.
- Spinal nerve root tumors: Schwannomas in NF2, neurofibromas in NF1.
- Cognitive impairment is not associated with NF2.
- NF2 has a high prevalence of bilateral vestibular schwannomas and meningiomas.
Noonan Syndrome
- Clinical Features: Characterized by short stature, webbed neck, characteristic facial features (hypertelorism, downward eye slant, low-set ears), and pulmonic stenosis.
- Overlap with NF1:
- May have café-au-lait spots, sometimes more than six and larger than 5 mm, meeting NF1 diagnostic criteria in children.
- Café-au-lait macules may be darker than usual in NF1.
- Facial features of Noonan syndrome may also be seen in individuals with NF1.
- Genes: Due to mutations in several genes in the Ras signaling pathway, especially PTPN11.
References
"Neurofibromatosis type 1 (NF1): Pathogenesis, clinical features, and diagnosis." UpToDate. Retrieved from https://www.uptodate.com/contents/neurofibromatosis-type-1-nf1-pathogenesis-clinical-features-and-diagnosis.
Cite this: Cite this: ICNApedia contributors.Neurofibromatosis. ICNApedia, The Child Neurology Knowledge Environment. 25 November 2024. Available at: https://icnapedia.org/knowledgebase/articles/neurofibromatosis Accessed 25 November 2024.