
Aicardi-Goutieres syndrome (AGS) is a genetic encephalopathy characterized by the inappropriate induction of a type I interferon-mediated immune response and usually results in severe cognitive and physical morbidities. It is named after Jean Aicardi and Francois Goutières who first described the condition in 1984 [Aicardi J, Goutières F (1984) ] in patients who had presented with early onset encephalopathy, basal ganglia calcification, and persistent lymphocytosis in the cerebrospinal fluid. The condition has subsequently been shown to be both genotypically and clinically heterogenous.
Clinical features
There are two distinct forms of the syndrome:
- Early onset
- severe
- affects about 20 percent of all infants who have AGS
- symptoms present at birth with hepatomegaly, splenomegaly, elevated liver enzymes
- irritability & poor feeding (mimicking congenital viral infection)
- late-onset form
- less impact upon neurological function
- symptoms after the first weeks or months of normal development
- progressive decline in head growth
- muscle weakness & spasticity
- moderate to severe cognitive and developmental delay
- Symptoms last for several months, and include irritability, inconsolable crying, intermittent fever, seizures, and loss of developmental skills
- Skin manifestations include puffy swelling on the fingers, toes, and ears that resemble chilblains, acrocyanosis, periungual erythema, or necrotic
- lesions of the toes, fingers, and outer helix
- startle reaction to sudden noise
- no further worsening of the disease once the symptoms lessen
- 50% - 75% of patients with AGS develop focal and generalized seizures, which are sometimes refractory to treatment [Ramantani et al., 2014].
However the clinical features can be quite heterogenous. The neurological dysfunction is not always severe and need not even be present at all. Other features originally described including microcephaly, intracranial calcification, white matter changes and CSF lymphocytosis are not always present [].
Genetics
AGS is a genetically heterogeneous disease. Most cases of AGS are inherited in an autosomal recessive manner. However it can also be autosomal dominant with heterozygous mutations in TREX, ADAR, or IFIH1 genes. In some cases AGS-associated mutations show incomplete penetrance with children in the same family with the same mutations showing markedly different neurological and developmental outcomes [Crow YJ et al., 2015]. To date AGS has been associated with mutations of seven different genes.
{tabulizer:include style[gr.alterora.elemental_2_grey.css] id[tab_mCJ6uOOecl]}
| Type | OMIM | Gene | Locus | Frequency | Inheritance | Phenotypes | Protein Function |
| AGS1 | 225750 | TREX1 | 3p21.31 | 23% (1% dominant) | AR (rare AD cases reported) | AGS, FCL, RVCL, SLE | 3′-5′ exonuclease with preference for ssDNA |
| AGS2 | 610181 | RNASEH2B | 13q14.3 | 36% | AR | AGS | Non-catalytic component of RNase H2 complex |
| AGS3 | 610329 | RNASEH2C | 11q13.1 | 12% | AR | AGS | Non-catalytic component of RNase H2 complex |
| AGS4 | 610333 | RNASEH2A | 19p13.2 | 5% | AR | AGS | Catalytic component of RNase H2 complex |
| Acts on RNA portion of RNA/DNA hybrids and removes ribonuceotides embedded in DNA | |||||||
| AGS5 | 612952 | SAMHD1 | 20q11.23 | 13% | AR | AGS, FCL | dNTP triphosphohydrolase triphosphatase |
| AGS6 | 615010 | ADAR | 1q21.3 | 7% (1% dominant) | AR, AD | AGS, DSH | Hydrolytic deamination of adenosine to inosine in dsRNA |
| AGS7 | 615846 | IFIH1 | 2q24 | 3% (all dominant) | AD | AGS |
Pathophysiology
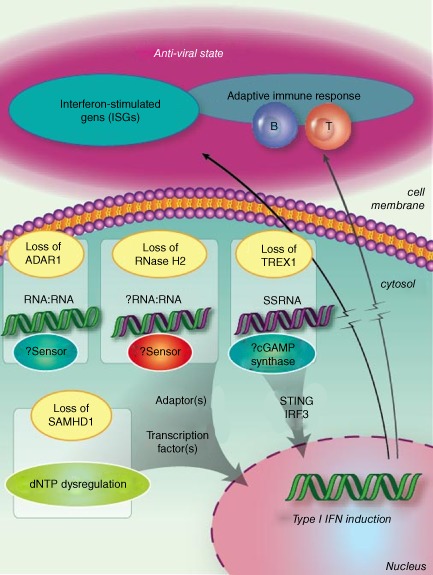
Both AGS and many placentally acquired viral infections are characterized by the production of high levels of IFNα. Astrocyte-specific chronic overproduction of IFNα in transgenic mice has been shown to recapitulate the neuropathologic findings seen in AGS [Akwa Y et al., 1998]. AGS shows a phenotypic overlap with some aspects of systemic lupus erythematosus (SLE) where an equivalent type I interferon-mediated innate immune response is triggered by self nucleic acids. Current experimental evidence suggests that the nucleases defective in AGS are involved in removing endogenously produced nucleic acid species, and that a failure of this removal results in activation of the immune system [Crow YJ et al., 2009].
The actual pathomechanisms that lead to CNS damage in AGS patients are unknown. In a mouse model transgenic for a glial fibrillary acidic protein-interferon-alpha fusion protein (GFAP-IFN-α mice) where IFN α is expressed in an astrocyte-restricted fashion, transgenic animals develop a progressive inflammatory encephalopathy with neuropathologic features similar to AGS suggesting a neurotoxic role of IFNα in the developing brain [Akwa Y et al., 1998].
Interferonopathies comprise an expanding group of monogenic diseases characterised by disturbance of the homeostatic control of interferon (IFN)-mediated immune responses [].
Diagnosis
- AGS is difficult to diagnose since many of the symptoms are similar to those of other disorders
- Diagnosis is based on the clinical symptoms of the disease
- Radiological abnormalities include basal ganglia calcification (usually bilateral) involving globus pallidus, putamen, caudate nucleus, and dentate nuclei. It may also extend to the periventricular white matter; increased signal on T2 weighted brain MRI particularly in the frontotemporal region along with cerebral atrophy [La Plana R et al., 2016] [].
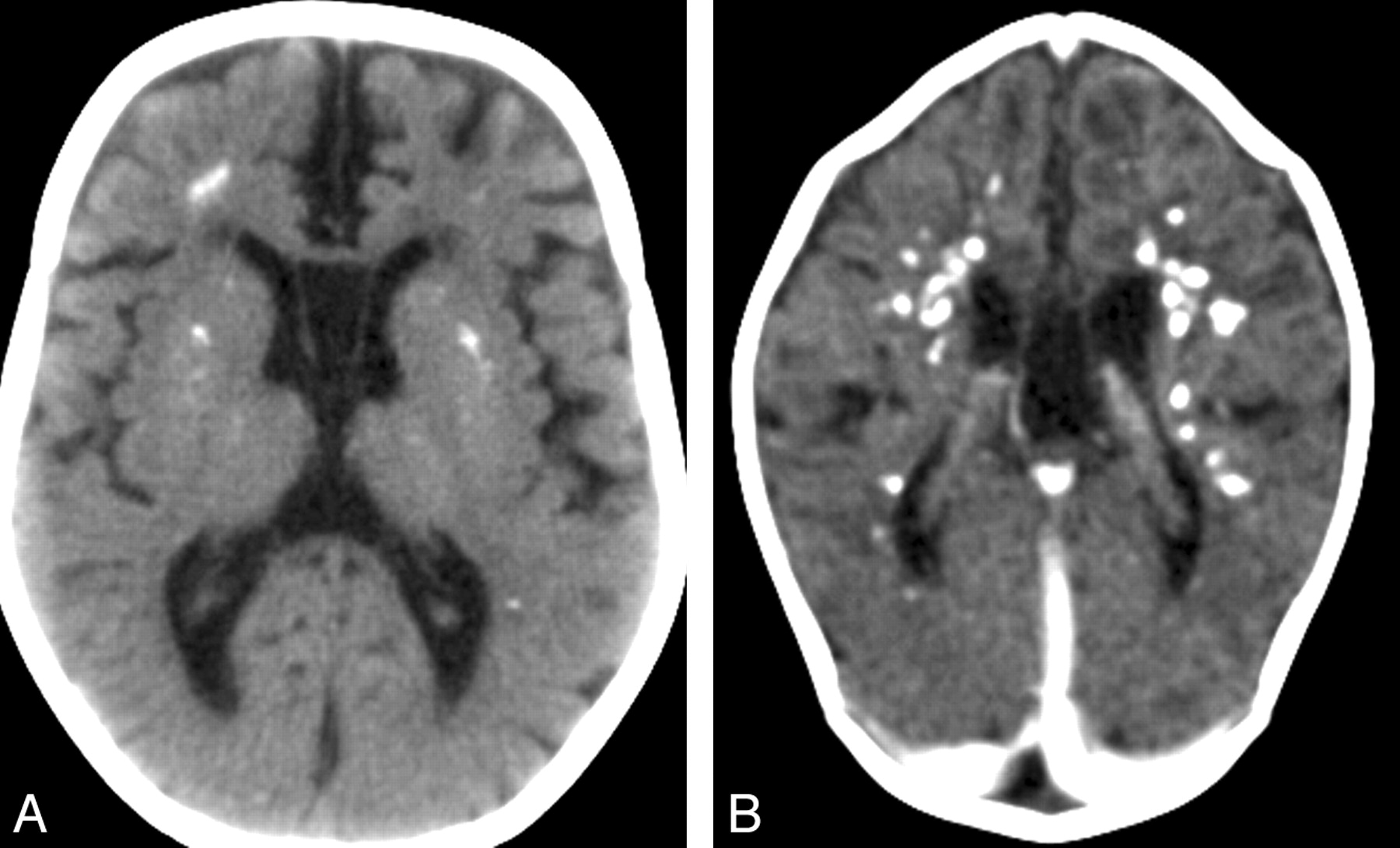 Cerebral calcifications. A, Axial nonenhanced CT image showing numerous punctuate calcifications within the basal ganglia and the cerebral white matter B, Contrast-enhanced CT scan showing large calcifications in the white matter.Atrophy, microcephaly, and areas of hypoattenuation in the periventricular white matter are also shown.
Cerebral calcifications. A, Axial nonenhanced CT image showing numerous punctuate calcifications within the basal ganglia and the cerebral white matter B, Contrast-enhanced CT scan showing large calcifications in the white matter.Atrophy, microcephaly, and areas of hypoattenuation in the periventricular white matter are also shown.
- Cerebrospinal fluid (CSF) shows increased levels of lymphocytes indicating chronic lymphocytosis in the absence of evidence of infection.
- Raised interferon γ in CSF
Treatment
-
Treatment in the early stages of the disease might result in attenuation of the associated inflammation and consequent tissue damage
-
Treatment may be discontinued once the subacute encephalopathic period subsides
-
Treatment may have to be continued beyond the subacute encephalopathic phase in cases where chilblains are a particular problem
-
immune-modulating therapies have been tried empirically including prednisone with azathioprine, intravenous (i.v.) methylprednisolone with i.v. immunoglobulin (IVIG), methylprednisolone and IVIG alone with variable results.
-
Anti-interferon alpha antibody therapy: blocking interferon alpha activity using monoclonal antibodies is currently under consideration and clinical trials are currently underway in SLE [Yao Y et al., 2009]
-
Reverse transcriptase inhibitors (RTIs) (compounds that can potentially disrupt the replication cycle of both exogenous retroviruses and endogenous retro-elements) are of potential interest in treating AGS. RTIs are already prescribed in children and adults with HIV-1 infection [Crow YJ et al., 2014]
-
Agents that deplete B cells are also potential therapeutic agents since AGS is associated with lymphocytes and autoantibody production
-
Other agents of interest include drugs that target autoreactive T cells such as Mycophenolate Mofetil
References
Aicardi J, Goutières F (1984) A progressive familial encephalopathy in infancy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis. Ann Neurol 15 (1):49-54. DOI: 10.1002/ana.410150109 PMID: 6712192.
Akwa Y, Hassett DE, Eloranta ML, Sandberg K, Masliah E, Powell H | display-authors=etal (1998) Transgenic expression of IFN-alpha in the central nervous system of mice protects against lethal neurotropic viral infection but induces inflammation and neurodegeneration. J Immunol 161 (9):5016-26. PMID: 9794439.
. Aicardi-Goutieres syndrome and related phenotypes: linking nucleic acid metabolism with autoimmunity. Hum Mol Genet. 2009 Oct 15;18(R2):R130-6. doi: 10.1093/hmg/ddp293.
. Therapies in Aicardi-Goutières syndrome. Clin Exp Immunol. 2014 Jan;175(1):1-8. doi: 10.1111/cei.12115
Crow YJ, Chase DS, Lowenstein Schmidt J, Szynkiewicz M, Forte GM, Gornall HL | display-authors=etal (2015) Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am J Med Genet A 167A (2):296-312. DOI: 10.1002/ajmg.a.36887 PMID: 25604658.
. Genetic interferonopathies: An overview. Best Pract Res Clin Rheumatol. 2017 Aug;31(4):441-459. doi: 10.1016/j.berh.2017.12.002. Epub 2018 Feb 1.
. Neuroradiologic patterns and novel imaging findings in Aicardi-Goutières syndrome. Neurology. 2016 Jan 5;86(1):28-35. doi: 10.1212/WNL.0000000000002228. Epub 2015 Nov 18.
Ramantani G, Maillard LG, Bast T, Husain RA, Niggemann P, Kohlhase J | display-authors=etal (2014) Epilepsy in Aicardi-Goutières syndrome. Eur J Paediatr Neurol 18 (1):30-7. DOI: 10.1016/j.ejpn.2013.07.005 PMID: 24011626.
. Aicardi-Goutieres syndrome: neuroradiologic findings and follow-up. AJNR Am J Neuroradiol. 2009 Nov;30(10):1971-6. doi: 10.3174/ajnr.A1694. Epub 2009 Jul 23.
. Development of Potential Pharmacodynamic and Diagnostic Markers for Anti-IFN-α Monoclonal Antibody Trials in Systemic Lupus Erythematosus. Hum Genomics Proteomics. 2009 Nov 17;2009:374312. doi: 10.4061/2009/374312. [PMID: 20948567] [PMCID: 2950308] [DOI: 10.4061/2009/374312]
