Introduction
Beta-propeller protein-associated neurodegeneration (BPAN) is a rare form of neurodegeneration with brain iron accumulation (NBIA). It was first described by Haack et al. in 2012, initially termed static encephalopathy of childhood with neurodegeneration in adulthood (SENDA) . BPAN is characterized by static psychomotor retardation in childhood followed by progressive deterioration in adolescence or young adulthood, featuring progressive dystonia, parkinsonism, and dementia. Pathogenic mutations in the WDR45 gene are responsible for BPAN, which constitutes about 7% of NBIA disorders .
Clinical Spectrum
BPAN presents with delayed psychomotor development and intellectual disability from infancy or early childhood. A significant proportion (74.6%) of patients develop progressive cognitive decline in adolescence or early adulthood. Epileptic seizures occur in 67.7% of patients and range from focal to generalized seizures, often improving with age . Movement disorders, including dystonia (73.3%) and parkinsonism (60.3%), typically emerge during adolescence or early adulthood, leading to severe motor disability .
Rett-like Features
Rett-like features are observed in 28% of patients, characterized by developmental regression, loss of purposeful hand skills, stereotypic hand movements, and bruxism. Most cases do not meet all diagnostic criteria for Rett syndrome .
Additional Clinical Features
Spasticity, sleep disturbances, and ocular/visual defects are variably reported. Limb spasticity, a range of sleep disturbances, and various ocular/visual issues, including myopia, astigmatism, and optic atrophy, have been documented . Dysmorphic features are seen in some patients, with characteristics such as microcephaly, abnormal nasal bridge, and epicanthal folds . Neuropsychiatric symptoms, “happy demeanor,” and excessive drooling are other clinical features noted .
Imaging Findings
MRI findings consistent with iron deposition in the basal ganglia are seen in 90.2% of patients. Typical MRI features include hypointense signals in the substantia nigra and globus pallidus on T2-weighted images and a hyperintense “halo” on T1-weighted images . Additional findings include cerebral atrophy, cerebellar atrophy, delayed myelination, thin corpus callosum, and dilated ventricles.
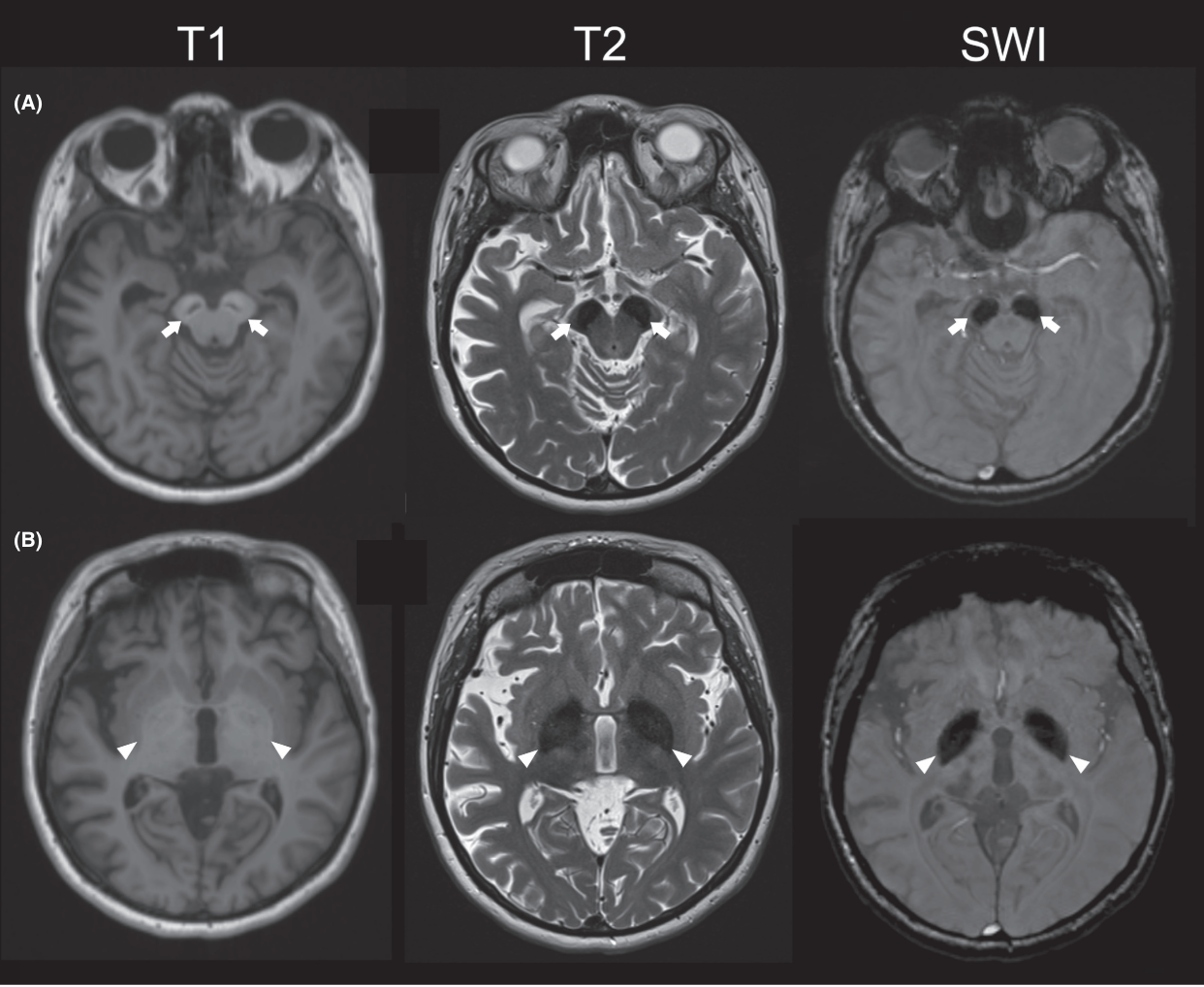 MRI findings in BPAN
MRI findings in BPAN
Pathophysiology
BPAN is caused by mutations in the WDR45 gene, encoding the WIPI-4 protein involved in autophagy. Mutations lead to lower autophagic activity and accumulation of aberrant autophagic structures, linking autophagy dysfunction to neurodegeneration .
Inheritance and Gender
BPAN is inherited in an X-linked dominant manner, with most cases resulting from de novo mutations. Females are predominantly affected due to reduced survival of male embryos with pathogenic mutations. The variability in disease severity is attributed to factors like the severity of the mutation and skewed X chromosome inactivation .
Treatment
Levodopa treatment improves clinical symptoms in 95.2% of BPAN patients with parkinsonism, though many experience side effects such as motor fluctuation and dyskinesias. Improvement in dystonia has also been reported in some cases .
Conclusion
BPAN presents a heterogeneous clinical spectrum. Early diagnosis and regular follow-up are crucial for managing the disease and providing genetic counseling. Understanding BPAN’s molecular mechanisms can aid in designing tailored therapies and provide insights into autophagy's role in neurodegeneration .
References
- Haack, T. B., P. Hogarth, M. C. Kruer, A. Gregory, T. Wieland, T. Schwarzmayr, et al. 2012. Exome sequencing reveals de novo WDR45 mutations causing a phenotypically distinct, X-linked dominant form of NBIA. Am. J. Hum. Genet. 91:1144–1149
- Stige KE, Gjerde IO, Houge G, Knappskog PM, Tzoulis C (2018) Beta-propeller protein-associated neurodegeneration: a case report and review of the literature. Clin Case Rep 6 (2):353-362. DOI: 10.1002/ccr3.1358 PMID: 29445477.