
Lafora disease is a rare genetic disorder involving glycogen metabolism disorder. It is inherited by autosomal recessive pattern presenting as a progressive myoclonus epilepsy and neurologic deterioration beginning in adolescence. It is characterized by Lafora bodies in tissues such as brain, skin, muscle, and liver.
Signs and Symptoms
- Onset of symptoms between 8-19 years, peak at 14-16 years; rare early onset in children as young as 5 years
- Initial symptoms: headaches, learning difficulties, and seizures
- Most common seizures: myoclonic seizures (jerking or shaking movements)
- Other seizure types:
- Tonic-clonic (muscle stiffening followed by jerking)
- Absence (staring spells)
- Atonic (body becomes limp)
- Complex partial (staring spells with jerking or repetitive movements)
- Focal occipital (blindness or hallucinations)
- Seizures worsen over time, potentially leading to status epilepticus (prolonged or back-to-back seizures)
- Motor symptoms: difficulty with balance, walking, coordination, and spasticity
- Cognitive and psychiatric symptoms: difficulty speaking, behavioral changes, depression, apathy, and progressive dementia
- Disease progression: intractable myoclonus, loss of physical functions, severe cognitive decline
- Prognosis: Within 6 years, around half lose the ability to move voluntarily or interact cognitively; 50% survive more than 11 years after symptom onset
Causes
- Caused by mutations in EPM2A or EPM2B (NHLRC1) genes
- EPM2A encodes laforin, EPM2B encodes malin; the malin-laforin complex regulates glycogen elongation
- Uncontrolled elongation leads to "Lafora bodies," abnormal glycogen particles damaging cells
- Lafora bodies accumulate in the nervous system, muscle, liver, and skin, causing symptoms
- Inheritance: Autosomal recessive pattern
- Both parents must carry the mutated gene for a 25% chance of an affected child per pregnancy
Affected Populations
- Equally affects adolescent males and females
- Higher frequencies in populations from the Mediterranean, Northern Africa, India, and Pakistan
- Estimated prevalence: 4 per 1,000,000 people (may be underestimated)
Disorders with Similar Symptoms
-
Juvenile Myoclonic Epilepsy (JME)
- Similar seizures but no progression, cognitive or motor deterioration
- Genetic cause rarely identified
-
Unverricht-Lundborg Disease (EPM1)
- Stimulus-sensitive myoclonus and tonic-clonic seizures
- Ataxia, tremor, dysarthria, emotional sensitivity, depression, cognitive decline
- Slower progression, earlier onset, no Lafora bodies on skin biopsy
-
Myoclonic Epilepsy with Ragged Red Fibers (MERRF)
- Multisystem mitochondrial disorder
- Symptoms: myoclonus, generalized epilepsy, ataxia, myopathy, dementia, short stature, optic atrophy, hearing loss, cardiomyopathy, peripheral neuropathy
-
Subacute Sclerosing Panencephalitis (SSPE)
- Progressive neurological disorder due to measles infection
- Cognitive decline, behavioral changes, myoclonic jerks, seizures, vision issues
Diagnosis
- Initial symptoms: focal or myoclonic seizures
- Diagnostic confirmation: skin biopsy showing Lafora bodies, molecular genetic testing for EPM2A or EPM2B mutations
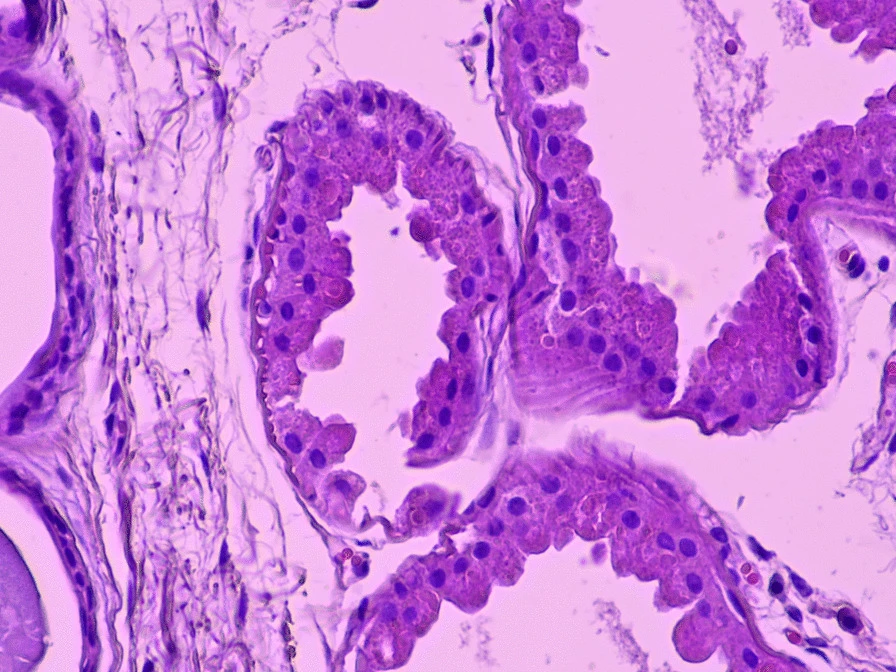
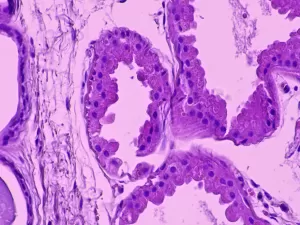 Axillary skin biopsy showing myoepithelial cells that contain polyglucosal (Lafora bodies) as well as chronic inflammatory infiltrates. The results of the biopsy reveal Periodic Acid-Schiff (PAS)+ polyglycan inclusions (Lafora bodies). Zeka, N et al. Lafora disease: a case report. J Med Case Reports 16, 360 (2022).
Axillary skin biopsy showing myoepithelial cells that contain polyglucosal (Lafora bodies) as well as chronic inflammatory infiltrates. The results of the biopsy reveal Periodic Acid-Schiff (PAS)+ polyglycan inclusions (Lafora bodies). Zeka, N et al. Lafora disease: a case report. J Med Case Reports 16, 360 (2022).
Clinical Testing and Work-Up
- Electroencephalogram (EEG) or magnetic resonance imaging (MRI) to screen for seizures and other disorders
Standard Therapies
- No cure; treatment focuses on symptom management
- Medical team: neurologist, geneticist, physical therapist, occupational therapist
- Symptom management: anti-seizure medications (valproic acid, perampanel), benzodiazepines
- Emergency treatment for myoclonic clusters or status epilepticus
- Genetic counseling and psychosocial support recommended
Clinical Trials and Studies
- Metformin (typically used for type 2 diabetes) shows promise in early treatment of Lafora disease
- Designated an orphan drug by the U.S. FDA, not yet approved for Lafora disease treatment
References
Nissenkorn A, Kluger G, Schubert-Bast S, et al. Perampanel as precision therapy in rare genetic epilepsies. Epilepsia. Published online February 2, 2023. doi:10.1111/epi.17530
Burgos DF, Machío-Castello M, Iglesias-Cabeza N, et al. Early treatment with metformin improves neurological outcomes in Lafora disease. Neurotherapeutics. Published online October 27, 2022. doi:10.1007/s13311-022-01304-w
Zeka N, Zogaj L, Gerguri A, et al. Lafora disease: a case report. J Med Case Rep. 2022;16(1):360. doi:10.1186/s13256-022-03537-x
Mitra S, Gumusgoz E, Minassian BA. Lafora disease: Current biology and therapeutic approaches. Rev Neurol. 2022;178(4):315-325. doi:10.1016/j.neurol.2021.06.006
Pondrelli F, Muccioli L, Licchetta L, et al. Natural history of Lafora disease: a prognostic systematic review and individual participant data meta-analysis. Orphanet J Rare Dis. 2021;16(1):362. doi:10.1186/s13023-021-01989-w
Markussen KH, Macedo JKA, Machío M, et al. The 6th International Lafora Epilepsy Workshop: Advances in the search for a cure. Epilepsy Behav. 2021;119:107975. doi:10.1016/j.yebeh.2021.107975
Orsini A, Valetto A, Bertini V, et al. The best evidence for progressive myoclonic epilepsy: A pathway to precision therapy. Seizure. 2019;71:247-257. doi:10.1016/j.seizure.2019.08.012
Bisulli F, Muccioli L, d’Orsi G, et al. Treatment with metformin in twelve patients with Lafora disease. Orphanet J Rare Dis. 2019;14(1):149. doi:10.1186/s13023-019-1132-3
Nitschke F, Ahonen SJ, Nitschke S, Mitra S, Minassian BA. Lafora disease – from pathogenesis to treatment strategies. Nat Rev Neurol. 2018;14(10):606-617. doi:10.1038/s41582-018-0057-0
Turnbull J, Tiberia E, Striano P, et al. Lafora disease. Epileptic Disord. 2016;18(S2):38-62. doi:10.1684/epd.2016.0842
Goldsmith D, Minassian BA. Efficacy and tolerability of perampanel in ten patients with Lafora disease. Epilepsy Behav. 2016;62:132-135. doi:10.1016/j.yebeh.2016.06.041
Resources
Ibrahim F, Murr N. Lafora Disease. StatPearls Publishing; 2022. https://www.ncbi.nlm.nih.gov/books/NBK482229/ Accessed March 13, 2023.
Velez-Bartolomei F, Lee C, Enns G. MERRF. 2003 Jun 3 [Updated 2021 Jan 7]. In: Adam MP, Mirzaa GM, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1520/ Accessed May 31, 2023.
Lehesjoki AE, Kälviäinen R. Progressive Myoclonic Epilepsy Type 1. 2004 Jun 24 [Updated 2020 Jul 2]. In: Adam MP, Mirzaa GM, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1142/ Accessed March 13, 2023.
Lafora disease. Genetic and Rare Diseases Information Center. https://rarediseases.info.nih.gov/diseases/8214/lafora-disease Accessed March 13, 2023.
NHL REPEAT-CONTAINING PROTEIN 1; NHLRC1. Online Mendelian Inheritance in Man (OMIM). Entry 608072. https://www.omim.org/entry/608072?search=nhlrc1&highlight=nhlrc1 Accessed May 31, 2023.
Lafora disease (Concept Id: C0751783). MedGen. National Center for Biotechnology Information. https://www.ncbi.nlm.nih.gov/medgen/C0751783 Accessed May 31, 2023.
Entry – #254770 – EPILEPSY, MYOCLONIC JUVENILE; EJM. Online Mendelian Inheritance in Man (OMIM). Entry 54770. https://omim.org/entry/254770 Accessed May 31, 2023.
